Spinal Disorders: Fundamentals of Diagnosis and Treatment Part 68 ppsx
Bạn đang xem bản rút gọn của tài liệu. Xem và tải ngay bản đầy đủ của tài liệu tại đây (349.69 KB, 10 trang )
24
Neuromuscular Scoliosis
Jean A. Ouellet, Vincent Arlet
Core Messages
✔
Kyphoscoliosis is a synonym for neuromuscular
scoliosis, in contrast to lordoscoliosis, which is a
synonym for idiopathic scoliosis
✔
Hyperlordosis is also seen in neuromuscular
scoliosis
✔
Pelvic obliquity is pathognomonic for neuro-
muscular scoliosis
✔
Spinal deformities in neuromuscular patients
tend to be severe and progressive in both coro-
nal and sagittal planes
✔
Surgical management of patients with neuro-
muscular scoliosis is associated with greater
morbidity as they can have severe comorbid
medical problems
✔
Duchenne muscular dystrophy and Friedreich’s
ataxia should always have a preoperative car-
diac assessment
✔
Preoperative pulmonary function of less than
35% of the predicted value indicates postopera-
tive ventilatory support and dependency, which
may put the surgical indications in question
✔
Maximizing hemostasis with adjuvant con-
trolled hypotension, cell savers, hemostatic
agents and excellent vascular access is impera-
tive since intraoperative bleeding can be signif-
icant (up to two times blood volume)
✔
Spinal fixation may be complicated and prone
to failure since bone is weakened by disuse,
osteopenia and antiepileptic drugs
✔
Achieving spinal balance in both the coronal
and sagittal planes is even more critical as
patients with neuromuscular scoliosis typically
do not have the innate ability to compensate
and balance themselves postoperatively
✔
Fusion often extends to the pelvis; thus a good
understanding of different pelvic-lumbosacral
fixations is mandatory
✔
Never extend a fusion down to the pelvis in a
patient relying on a mobile lumbosacral junc-
tion for his or her ambulation, even in the pres-
ence of pelvic obliquity
✔
If the curve <40° and the pelvic obliquity < 10°,
one can stop the fusion at L5; if these are
greater then the fusion should be extended to
the pelvis
Epidemiology
Neuromuscular scoliosis
embodies a heterogeneous
group of patients
Scoliosis in the presence of a neuromuscular disorder (NMD) behaves entirely
differently from the more predictable idiopathic scoliosis. Depending on the
underlying NMD, the prevalence of scoliosis is also different. Having a better
understanding of these disorders facilitates the management of their associated
spinal deformities (
Table 1).
Treatment must be
individualized for each
underlying diagnosis
One must appreciate that the heading of neuromuscular scoliosis encom-
passes a large variety of different NMD pathologies. These disorders can present
either early or later in life. They can be acquired by means of postinfectious or
post-traumatic events, or they can be genetic disorders affecting genes that code
for the proteins in nerve cells or in muscle cells, leading to malfunction of the
neurological or muscular systems. They can also be secondary to brain or spinal
cord insults or disease. The majority of these disorders present in different sever-
Spinal Deformities and Malformations Section 663
a b cd
e
f
Case Introduction
A 4-year-old boy with Duchenne muscular dystrophy had been followed at
the neuromuscular clinic at regular intervals to monitor respiratory status
and general development. On initial screening, spine X-ray did not demon-
strate any spinal deformity (a, b). At the age of 6, spinal asymmetry was
noted and a 10° scoliosis documented. By the age of 10, the curve had pro-
gressed to 48° (
c). Respiratory functions were 35% of expected and
deemed amenable to spinal surgery with moderate perioperative risk. The
patient had a classic segmental posterior spinal fusion using sublaminar
wiring from T2 to L5 (
d). A decision was made to fuse to L5 and not fuse to
the pelvis considering that his pelvic obliquity was minimal <10° and flexi-
ble (
e, f). By doing so the risk of pseudoarthrosis across the lumbosacral
junction was minimized. Being a male and non-ambulator the fusion could
have been extended to the pelvis to prevent the possibility of progressive
pelvic obliquity. In girls that perform self-catherization, fusing to the pelvis
often leads to loss of independence of self-care. The second contentious
decision was that no anterior spinal fusion was done due to the fear that he
would not tolerate the extended surgery. Fusing the spine at such a young
age poses a risk of the patient developing a crankshaft deformity; however,
considering that he had passed his peak growth velocity, this risk was mini-
mal. Furthermore any decisions must take into account his truncated life
expectancy. Of note is that the rods were inappropriately contoured lack-
ing lumbar lordosis to achieve an adequate sagittal balance.
Table 1. Characteristics of neuromuscular disorders associated with scoliosis [15, 34, 47]
Disease
(incidence)
Onset
(years)
Inheritance Life
expectancy
(years)
Presentation Progression of
weakness
Loss of
ambula-
tion (years)
Muscular dystrophies
Duchenne
(1:4000 male
births)
1.5–4 XR 20±4 Proximal muscle weakness, lower
weaker than upper limbs, extensor
weaker than flexor, muscles of
heart and respiratory system
Rapid decline
from 5 to
13 years, slower
after 14
10 ±2.5
Becker
(4:100 000
male births)
8.5±8.5 XR 23–89 Distribution similar to Duchenne Slow decline 25–58
664 Section Spinal Deformities and Malformations
Table 1. (Cont.)
Disease
(incidence)
Onset
(years)
Inheritance Life
expectancy
(years)
Presentation Progression of
weakness
Loss of
ambula-
tion (years)
Muscular dystrophies
Limb girdle
(incidence
cannot be
estimated)
9±4 AR (expAD) variable distribution similar to Duchenne
and Becker except no difference of
extensor and flexor
rapid loss 75% by
age 20
Myotonic (AKA
Steinert’s)
(1:20000
births)
23±13 AD variable
(dependent
on arrhyth-
mias)
facial weakness notice first, ptosis,
generalized weakness of voluntary
muscles of limbs, distal muscle
weakness, and the neck, facial, and
diaphragm muscles, and intercos-
tals. Develops heart blocks, unable
to release grasp
slow loss late in life
if ever
Congenital
myotonic
at birth AR variable
(% neona-
tal death)
severe weakness, floppy baby,
require ventilation and nutrition
supplement as infant, moderate
mental retardation
may never
reach
ambula-
tion
Arthrogryposis
(1:3 000 births)
at birth non-genetic
fetal akine-
sia, 30% AR
normal
(50% neo-
natal death
when CNS)
focal weakness in presence of
severe joint contractures: classic
hands, wrists, elbows, shoulders,
hips, feet and knees. Severe cases,
all joints including jaw and spine
static; may pro-
gress with dis-
use, atrophy
may be present,
and muscles or
muscle groups
may be absent
variable
spinal muscular atrophy (1 : 6000 births)
Type I (acute
infantile, acute
Werdnig-Hoff-
mann disease)
0–0.5 AR 1.5 (50 %
die before
2years)
severe generalized muscle weak-
ness leading to feeding and breath-
ing failures, unable to sit
never
ambulate
Type II (chronic
Werdnig-Hoff-
mann diseases)
2 30–40 proximal muscle weakness, lower
weaker than upper limbs, extensor
weaker than flexor, sits but diffi-
culty walking if able
progression
variable
early loss
Type III (Kugel-
berg-Welander
diseases)
23±19 normal proximal muscle weakness, no dif-
ference between lower and upper
or flexor and extensor
slow loss very late if
any
Poliomyelitis
(prevalence in
2003: 623 cases
worldwide)
variable acquired
(Nigeria,
India, Paki-
stan, Afgha-
nistan,
Egypt)
normal
(may
require
respiratory
support)
prodrome: fever 5–7 days before
headache, stiff neck, paraspinal
muscle weakness, asymmetrical
peripheral weakness (only on one
side or worse on one side), distribu-
tion depends on level of cord
involvement, abnormal sensations
with hypersensitivity
rapid onset
progresses to
paralysis, per-
manent or tran-
sient with pos-
sible mild
delayed regres-
sion
variable
depen-
dent on
severity,
subclini-
cal, non-
paralytic,
paralytic
Hereditary motor sensory neuropathy
Charcot-Marie-
Tooth (1:2 500
births)
13±14 AD relatively
normal
distal muscle weakness, no differ-
ence upper vs lower, nor flexor vs
extensors
slow loss later if any
Cerebral palsy
(2:1000 births)
at birth acquired
brain insult
in utero/peri-
natally, post-
infectious
variable
(dependent
on mobility;
non-sitter:
30; sitter:
46; ambula-
tor: 62)
spastic (50%): stiff, difficult move-
ment
dyskinetic/athetoid (20%): involun-
tary uncontrolled movement
ataxic (rare): poor coordination and
balance
mixed (30%): combination of these
types
hypotonia may
develop into
spasticity
variable
Spinocerebellar dysfunction
Friedreich’s
ataxia (1:22000
births)
10±5 AR early
adulthood
38 ±14
(cardiac)
initially difficulty walking, ataxia,
then spreading to arms then trunk,
muscle weakness, muscle wasting:
feet, leg, hands, loss of sensation
over time, nystagmus, cardiomyop-
athy, myocardial fibrosis
slow progres-
sive
15–20
years after
diagnosis
Neuromuscular Scoliosis Chapter 24 665
ities: from mild, to moderate, to severe forms. They may result in minimal clini-
cal manifestation or they can result in lethal disease in early infancy. An overview
of these disorders with their clinical presentations, their incidence and their
functional impact is given in
Table 1.
Disease Specific Spinal Deformity
As part of a review of 547 individuals with different NMDs, the Rehabilitation
Research and Training Center (RRTC/NMD) found that the overall incidence of
Spinal deformity is frequent
and severe in rapidly
progressive NMD
spinal deformity was elevated (60–80%) in patients with rapidly progressive
NMD who presented before skeletal maturity [41], while in slowly progressive
NMD the incidence of scoliosis was relatively low (only 32%). In the patients with
rapidly progressive NMD, the incidence and severity of the scoliosis increased
with disease duration and years of wheelchair dependency, with a high incidence
of pulmonary complications and decreased pulmonary function. In contrast, in
patients with slowly progressive NMD, the presence of spinal deformity showed
no relationship between disease duration and length of wheelchair dependency.
The scoliosis of these patients was often mild to moderate and usually non-pro-
gressive. There was, however, a significant association between the number of
pulmonary complications and disease duration in those patients with spinal
deformity who also had significantly lower vital capacities. One must keep in
mind that these are general guidelines and do not imply a cause to effect relation-
ship between specific disease and the development of scoliosis.
Duchenne patients are
likely to develop scoliosis
For example, in Duchenne muscular dystrophy (DMD), there is a progressive
increase in incidence of scoliosis up to the age of 20 years (
Case Introduction).
The incidence increases significantly once patients are wheelchair dependent,
especially after 3 years, when the incidence is close to 60%. Thirty-five percent of
patients have spinal deformity before the age of 8 years, and 90% do so by the age
of 20 years [15]. The incidence increases greatly between the ages of 13 and
15 years, which correspond closely with the adolescent growth spurt in boys.
In contrast, in patients with Becker’s muscular dystrophy,only13%hadscoli-
osis with mild non-progressive curves. Patients with hereditary motor sensory
neuropathy (HMSN, Charcot-Marie-Tooth disease) had a 25% incidence of spi-
nal deformity, of whom 15% had scoliosis and 10% had kyphoscoliosis. In
patients with Friedreich’s ataxia, the incidence of scoliosis was almost 100%,
compared to only 32% in those with other types of hereditary spinal cerebellar
ataxia (HSCA). Patients with infantile onset spinal muscular atrophy (SMA) had
a 78% incidence of scoliosis while juveniles and adults with SMA onset had only
8% incidence. Spinal deformity in the congenital myopathies occurred primarily
in the individuals with congenital muscular dystrophy (36%). Thirty-five percent
of patients with facioscapulohumeral dystrophy had spinal deformity, of whom
15% had scoliosis alone. The incidence of spinal deformity in limb girdle syn-
drome also depended on the type. Individuals with the childhood onset type had
a 44% incidence while those with the late onset and pelvofemoral types had only
a 6% incidence. There was a marked difference in the incidence of spinal defor-
mity between congenital myotonic muscular dystrophy (MMD) and non-con-
genital MMD. Forty-seven percent of the former had scoliosis as compared to
15% of the latter.
Ninety percent of myelodys-
plasia patients with a T10
level will develop a spinal
deformity
With respect to patients with myelodysplasia, the prevalence will vary
depending on their functional level: 90% of patients with a complete T10 level
will develop a coronal or sagittal spinal deformity, while only 5% of patients with
an L5 level will develop a spinal deformity [20].
The overall incidence of spinal deformity varies depending on the underlying
NMD, but it also varies according to the severity of the underlying NMD
666 Section Spinal Deformities and Malformations
Table 2. Prevalence of spinal deformities in neuromuscular diseases
Diagnosis Percentage
a
Cerebral palsy 25
Poliomyelitis 17–80
Myelodysplasia 60
Spinal muscular atrophy 67
Friedreich’s ataxia 80
Duchenne muscular dystrophy 90
Spinal cord injury (traumatic before 10 years of age) 100
a
Based on data by J.E. Lonstein, Department of Orthopedics, University of Minnesota, Twin
Cities Spine Center, Minneapolis
(Table 2). In general, the greater the neuromuscular involvement, the greater the
likelihood of having a spinal deformity and the greater the deformity will be.
Pathogenesis
The pathophysiology of neurogenic spinal deformities remains unclear. It seems
logical to assume that the “collapsing kyphoscoliosis” is secondary to muscle
weakness and yet the same deformity is seen in patients with spasticity. The clas-
sical spinal deformities encountered in NMD consist of:
scoliosis
kyphosis
kyphoscoliosis
lumbar hyperlordosis
pelvic obliquity
Pelvic obliquity is an
associated spinal deformity
Pelvic obliquity should be considered as an associated “spinal” neurogenic
deformity. All of these deformities canbe present with any of the different NMDs,
making it difficult to draw any conclusion about the pathogenesis of neuromus-
cular scoliosis. Furthermore there is no association between etiology, pattern of
weakness, and curve pattern. There are factors that influence the development of
certain deformities. For example, the development of scoliosis is influenced by
the following factors:
age of onset of NMD
ambulation status
severity and rapidity of the progression of the weakness
This is particularly true for patients with Duchenne muscular dystrophy. Close to
90% of them will develop scoliosis as their weakness progresses quickly, and it
occurs prior to cessation of growth coupled with loss of ambulation at an early
age. However, these factors do not always lead to a deformity, such as in patients
with amyotrophic lateral sclerosis, which is a very rapid progressive NMD and
yet only 1% develop scoliosis.
Classification
The classic patient we think of having neuromuscular scoliosis has either cerebral
palsy (upper motor neuron lesions) or Duchenne muscular dystrophy (peripheral
muscular disease) [4]. These two etiologies are representative of the two main
types of neuromuscular scoliosis. The Scoliosis Research Society has classified
neuromuscular scoliosis into neuropathic types and myopathic types (
Table 3
).
Neuromuscular Scoliosis Chapter 24 667
Table 3. Classification of neuromuscular scoliosis
Neuropathic conditions Myopathic conditions
Upper motor neuron
cerebral palsy
syringomyelia
spinal cord injury
Lower motor neuron
poliomyelitis
spinal muscular atrophy
Mixed upper and lower motor neuron
myelodysplasia (spina bifida)
spinal trauma
Spinocerebellar dysfunction
Friedreich’s ataxia
Hereditary motor sensory neuropathy
Charcot-Marie-Tooth
Muscular dystrophy
Duchenne and Becker
limb girdle
facioscapulohumeral
myotonic dystrophy
Arthrogryposis
Congenital myopathies
nemaline
central core disease
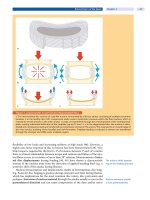
Lonstein et al. [22] classified the curve patterns of neuromuscular scoliosis in
patients with cerebral palsy and mental retardations into two large groups each
subdivided into two subgroups (
Fig. 1). The difference between the groups is the
presence (G-II) or absence (G-I) of pelvic obliquity, which has a clinical bearing
as to whether to include the pelvis in the spinal fusion.
Figure 1. Neuromuscular curve classification
Group I: double thoracic and lumbar curves, little pelvic obliquity, patient in balance. a Thoracic lumbar curve in balance;
b thoracic greater than lumbar curve, unbalanced. Group II: large lumbar or thoracolumbar curves, severe pelvic obliq-
uity, patient out of balance.
c Short fractional curve above sacrum; d extension of lumbar curve in sacrum (According to
Lonstein et al. [22]).
668 Section Spinal Deformities and Malformations
Clinical Presentation
History
As in any ailment, obtaining a detailed history is fundamental in the establish-
ment of the correct diagnosis of scoliosis. A thorough history should include:
perinatal history
development history
family history
A family history is required to assess the risk of a known etiology for the patient’s
spinal deformity. Clues suggestive for neuromuscular scoliosis are:
birth anoxia
delayed developmental milestone
acquired or familial neuropathies and/or myopathies
early onset (less than 7 years old)
painful scoliosis
Detailed perinatal history
and family history is
warranted if neuromuscular
scoliosis is suspected
The patient should be asked about maternal diabetes, specific bowel and bladder
functions, and muscle endurance since these insignificant details can lead to a
diagnosis of sacral agenesis or then again to that of a tethered cord. Subjective
complaints of patchy numbness and weakness must be elicited as well as symp-
toms consistent with radiculopathy, myelopathy, or recurrent headaches, which
mayallbesymptomsofasyringomyelia(
Table 4).
Table 4. Red flags for neuromuscular scoliosis
History:
early onset scoliosis: early, less than 7 years of age
painful scoliosis
headache
sensory or motor disturbances
bowel and bladder dysfunction
developmental delay, mental retardation
Physical examination:
Head & neck:
flaccid facies
poor head control
Skin:
neuroectodermal lesions: caf´eaulaitspots
spinal dysraphism: hairy patch, sacral dimples, midline birthmark
Spine:
long collapsing scoliosis
pelvic obliquity
kyphoscoliosis
lack of rotation
Neurology:
spasticity
muscle weakness, proximal girdle + Gower
peroneal muscular weakness
long track signs: clonus, Babinsky’s, hyperreflexia
hypotonia, hyporeflexia
patchy paresthesia
Musculoskeletal:
limb atrophy, different feet size
cavus feet
upper extremity posturing during running
loss of sitting balance
Charcot joints
non-ambulators
Neuromuscular Scoliosis Chapter 24 669
Physical Examination
Skin
Thedermismustbeinspectedforskinlesionssuchascaf´eaulaitspotsor axil-
lary freckles as these are associated with neurofibromatosis, which can have
intradural neuromas. Other neurocutaneous skin markings such as hairy
patches (
Fig. 2) or midline nevi (or vascular lesion) can also be superficial clues
to intradural pathologies.
Spine
Coronal imbalance
is frequent in
neuromuscular scoliosis
Neuromuscular scoliosis resembles a kyphoscoliotic deformity, in contrast to the
lordoscoliosis found in adolescent idiopathic scoliosis. Ky phosis is frequently
found as an associated spinal deformity in the neuromuscular patient as the
majority of them have “collapsing spine” secondary to muscular weakness or
deficient trunk control (
Case Study 1). Patients must be examined for both defor-
mities in the sitting and supine positions, giving us an immediate insight into the
overall rigidity of both deformities. Of note, hyperlordosis can also be seen in
neuromuscular scoliosis, leading to inability to sit properly.
Sagittal imbalance
with apical kyphosis
is also frequent
The combination of pelvic obliquity and scoliosis tends to lead to spinal
imbalance, resulting in abnormal pressure points. Patients with neuromuscular
scoliosis can develop pressure sores on the sacrum, the ischia, and the greater
trochanter and these should be looked for.
a
b
c
d
Figure 2. Clinical clues to neuromuscular scoliosis
a Eleven-year-old boy, idiopathic-like curve pattern, asymptomatic. On examination unilateral cavus foot with calf atro-
phy is noted.
b The patient presents with a myopathic scoliosis due to Charcot-Marie-Tooth disease. c Seven-year-old girl,
right thoracic curve, with overt neuroectodermal marker – hairy patch.
d The patient is diagnosed with diastematomye-
lia and tethered cord.
670 Section Spinal Deformities and Malformations
ab c
d
ef g
Case Study 1
A 12-year-old boy with congenital myopathy (a) presented at our neuromuscular clinic with his older brother (b), who
was also diagnosed with neuromuscular scoliosis. His brother had undergone a selective thoracic posterior spinal fusion
with Harrington rod 15 years earlier (
c). Over time the brother developed additional deformity above and below and
crankshaft deformity across the instrumented segment. The main concern of the younger brother was not to end up like
his older brother. The patient has severe coronal imbalance with a significant pelvic obliquity (
d, e). Surgical manage-
ment must address both the long classic C-shape neuromuscular scoliosis and the pelvic obliquity. The primary goal is
to achieve coronal and sagittal balance. Despite the relatively rigid upper thoracic deformity, correction was achieved by
posterior alone spinal surgery with a solid pelvic fixation comprising MW construct, pedicle screws above and below and
apical sublaminar wire to maximize apical translation (
f, g). The MW “segmental pelvic fixation” (see Fig. 5) allows (if
needed) for further pelvic correction by levering on the iliosacral screws in the up or down hemipelvis depending on
residual obliquity even after the cantilever maneuver has been done.
Neuromuscular Scoliosis Chapter 24 671
Pelvis and Hips
Hip contractures will
influence treatment
From a musculoskeletal examination point of view, one must assess the skeletal
appendages as well as the spine. A detailed examination of the hips particularly
looking for hip contracture is crucial as they influence sitting balance and in par-
ticular can induce pelvic obliquity (
Case Study 1). As there are many patients
with neuromuscular scoliosis who are wheelchair dependent, one must pay par-
ticular attention to the pelvis and its orientation in both the coronal (obliquity)
and sagittal plane (anteversion/retroversion).
If pelvic obliquity is present, one should assess whether its origin is:
suprapelvic
intrapelvic
infrapelvic [13]
Pelvic obliquity
is pathognomonic
for neuromuscular scoliosis
Suprapelvic obliquity is secondary to the spinal deformity itself. The scoliosis
drives the pelvis in its obliquity. Dubousset saw the pelvis as the 6th lumbar ver-
tebra and the pelvis being a simple extension of the scoliotic deformity resulting
in pelvic obliquity. In contrast, infrapelvic obliquity is secondary to hip contrac-
tures which result in pelvic obliquity. The contractures which drive the pelvic
obliquity tend to be abduction or adduction hip contractures. When both are
present in opposite hips one talks of windswept deformity of the hips, which typ-
ically results in significant pelvic obliquity.
NMD patients often develop
hip flexion contractures
In addition, as the majority of these patients are wheelchair dependent, they
develop hip flexion contractures. These may induce fixed or flexible sagittal spi-
nal deformity in the form of lumbar hyperlordosis. Orientation of the pelvis and
lumbar lordosis needs to be assessed as an anteverted pelvis or compensatory
hyperlordosis can indicate severe hip flexion contracture. These postoperatively
maybecomemuchmoreapparentasthepatientsarenolongerabletocompen-
sate with their flexible lumbar spine.
To differentiate between supra- and infrapelvic obliquity, the patient is placed
prone at the end of an examining table with the hips flexed over the edge of the
table (negating the flexion hip contractures). Then by abducting or adducting the
hips, the pelvis can be leveled in the infrapelvic obliquity, while for the suprapel-
vic obliquity the pelvis cannot be leveled by changing the position of the hips.
An understanding
of pelvic obliquity
is a key to treatment
Intrapelvic obliquity is secondary to morphological changesof the hemipelvi-
ses. This can be seen in asymmetrical myelomeningocele as the weaker side
develops less, resulting in bony architectural changes leading to ischial and ilium
hypoplasia. Pelvic X-rays are the only way to identify such pelvic obliquity.
Ambulatory Status and Mode of Ambulation
It is not enough to know if the patient is a:
walker
sitter (wheelchair bound)
non-sitter
Mode of ambulation
determines the extent
of instrumented fusion
In the walker, one must determine gait pattern and mode of ambulation. Certain
patients (myelodysplasia) need a mobile lumbosacral junction to ambulate as
they rely on pelvic thrust to propel their lower extremities to ambulate. Extend-
ing the fusion to the pelvis in this subpopulation would take away their ability to
ambulate. Even in the wheelchair-bound patient, a mobile lumbosacral junction
may be needed to perform self-catheterization. Thus, the decision to extend the
fusion to the pelvis must be done with careful consideration.
672 Section Spinal Deformities and Malformations